FDA验厂辅导
阅读:552779次
近年来SUNGO为国内众多制造商提供了FDA验厂的辅导和翻译陪审服务。这其中包括了FDA提前五天通知验厂的时间极端紧迫的案例,也包括了为连续两次验厂失败的制造商解除警告信并移除进口禁令的复杂度极高的案例。
PART 1 FDA工厂审查的概况:
FDA每年会对全球的医疗器械制造商进行抽样审查,作为其进行售后市场监管的主要途径之一。所有的审查都会由美国FDA的工作人员进行,不论这些人是什么族裔,他们都是美国籍,都代表了美国政府的利益。
近几年,在美国以外的国际市场,中国制造商的被抽样量一直稳居全球首位。目前中国在FDA的注册制造商约为4500家左右,每年抽查的概率在2-3%。通常FDA工厂审查会由1名审查官进行为期4天的现场审查。制造商无需支付任何审查费用。
PART 2 FDA工厂审查的直接后果:
大部分的中国制造商收到美国FDA的审查通知都会比较重视,基本上都会积极应对,动员内外部的力量和资源来确保审查顺利进行。当然也有部分制造商不了解审查可能会导致的结果,没有给予足够的重视,导致后面很被动的局面。
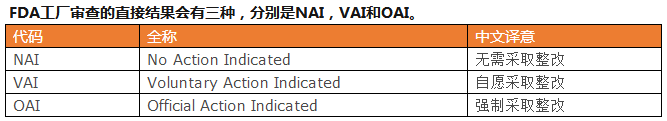
NAI表示在FDA工厂审查时,没有开出任何书面形式的不符合项(由于FDA的不合格报告表单的编号为483,FDA也将不符合简称“483”),也可以称为“零483”。
VAI表示在FDA工厂审查时,FDA审查官发现了工厂的管理系统有违背FDA的质量体系法规的内容,进而开具了书面形式的不符合项,也可以开具了“483”。“483”的个数可能是1个,也可能是20个或更多。只要工厂按照FDA的要求积极整改,提供充分的证据,都不会导致更多后果。
OAI表示在FDA工厂审查时,FDA审查官发现了工厂管理系统存在严重违背FDA的质量体系法规的内容,或者是没有能够按照FDA的要求对于VAI进行及时充分的整改,而开具的警告信(Warning Letter)。如果仅仅是开具了警告信而没有上Import Alert,制造商的产品依然可以出口,但是警告信会公布在FDA官网上,会影响美国客户对制造商的信心,必须尽快采取措施解除。
PART 3 FDA工厂审查的后果放大路径:
FDA的审查之所以让很多制造商觉得紧张,是因为稍有不慎,其结果的严重性可能会迅速放大,最终让制造商失去整个美国市场。其放大路径如下:
1. VAI没有按照FDA要求进行充分整改,会发展成为OAI;
2. OAI没有按照FDA要求及时响应,会被列入Import Alert;
3. 进入了Import Alert,企业的出口产品入境时会被自动没收(DWPE)。
由于FDA工厂审查导致的Import Alert要移除,通常都需要进行再次的工厂审查。除了再次的工厂审查之外,还有海量的证据需要随同请愿书提交FDA审查。从开始处理到最终完成Import Alert移除的过程,最高效的处理周期需要一年时间。当然伴随着的还有巨额的费用。

PART 4 避免严重后果的方法
面对可能的严重后果,预防永远是最有效和成本最低的途径。采取预防措施,我们的建议是:
1) 当您的产品进入美国市场之前,尽快建立起QSR820 体系。
2) 寻求第三方的专业机构进行辅导。
3) 寻求第三方的专业机构进行模拟审核。
4) 在收到FDA验厂通知时,尽快联系专业机构提供支持。
特别指出的是:有部分企业最终被开具警告信并进入Import Alert是由于FDA现场审查时候的翻译人员对于技术法规和公司的质量管理系统不清楚导致翻译不准确,最终导致审查员开具了很多不应该有的不符合。
深圳行知合一FDA验厂辅导优势 :
1) 辅导团队具备多年药品和医疗器械美资企业的法规工作经验;
2) 具有成功辅导多家企业通过FDA审核的经验;
3)落地化辅导经验,并能结合各不同企业硬件提供改善方案和策略;
4) 咨询师具有优秀的语言能力,担任陪审的任务会使得企业应对更轻松。

上一篇:
SQP审计
下一篇:
GSV C-TPAT反恐